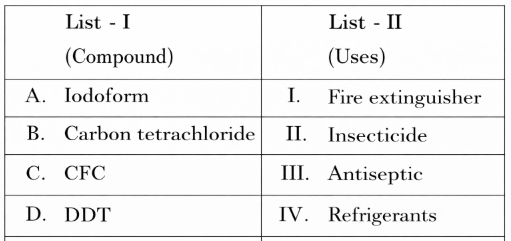
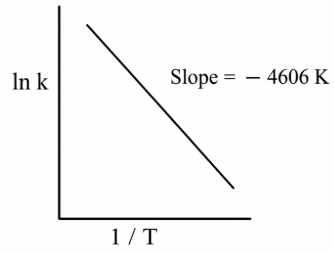
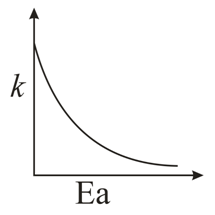
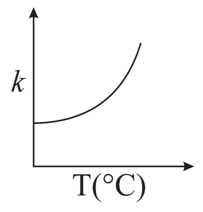
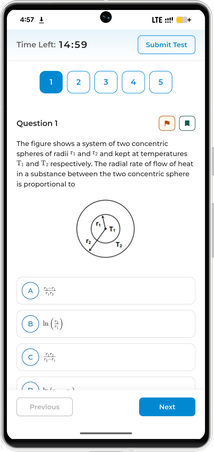
For any elementary reaction the Arrhenius equation gives the temperature dependence of the rate constant:
$$k = A\,e^{-E_a/RT}$$
where $$k$$ is the rate constant, $$A$$ is the frequency factor, $$E_a$$ is the activation energy, $$R$$ is the gas constant and $$T$$ is the absolute temperature in kelvin.
Taking two temperatures $$T_1$$ and $$T_2$$ for the same reaction, the ratio of the two rate constants is
$$\frac{k_2}{k_1}=e^{-E_a/R\,(1/T_2-1/T_1)} \;.$$
This may be rewritten, bringing the negative sign inside, as
$$\frac{k_2}{k_1}=e^{E_a/R\,(1/T_1-1/T_2)} \;.$$
We are told that for reaction A the rate doubles when the temperature is raised from $$T_1 = 300\ {\rm K}$$ to $$T_2 = 310\ {\rm K}$$, so $$k_2/k_1 = 2$$. Substituting these numbers, we have
$$2 = e^{E_{a,A}/R \,(1/300 - 1/310)} \;.$$
Taking natural logarithm on both sides,
$$\ln 2 = \frac{E_{a,A}}{R}\bigl(\, \frac{1}{300} - \frac{1}{310}\,\bigr).$$
Calculating the bracketed term first,
$$\frac{1}{300} = 0.0033333\quad{\rm K^{-1}},$$
$$\frac{1}{310} = 0.0032258\quad{\rm K^{-1}},$$
so
$$\frac{1}{300}-\frac{1}{310}=0.0033333-0.0032258 = 0.0001075\ {\rm K^{-1}}.$$
Now inserting this value,
$$\ln 2 = 0.693147 = \frac{E_{a,A}}{R}\times 0.0001075.$$
Hence
$$\frac{E_{a,A}}{R}= \frac{0.693147}{0.0001075}\approx 6.45\times 10^{3}\ {\rm K}.$$
For reaction B the problem states that its activation energy is twice that of reaction A, therefore
$$E_{a,B}=2E_{a,A}\quad\Longrightarrow\quad\frac{E_{a,B}}{R}=2\left(\frac{E_{a,A}}{R}\right)\approx 2\times6.45\times10^{3}=1.289\times10^{4}\ {\rm K}.$$
Let reaction B start at $$T_1 = 300\ {\rm K}$$ and let the temperature be raised to $$T_2 = 300+\Delta T$$. We want the rate to double again, so $$k_2/k_1 = 2$$. Using the same ratio form for reaction B, we write
$$2 = e^{E_{a,B}/R\,(1/300 - 1/T_2)}.$$
Taking the natural logarithm,
$$\ln 2 = \frac{E_{a,B}}{R}\Bigl(\frac{1}{300}-\frac{1}{T_2}\Bigr).$$
Now substitute the numerical value obtained for $$E_{a,B}/R$$:
$$0.693147 = 1.289\times10^{4}\Bigl(\frac{1}{300}-\frac{1}{T_2}\Bigr).$$
Dividing both sides by $$1.289\times10^{4}$$ gives
$$\frac{1}{300}-\frac{1}{T_2}= \frac{0.693147}{1.289\times10^{4}}\approx 5.378\times10^{-5}\ {\rm K^{-1}}.$$
Hence
$$\frac{1}{T_2}= \frac{1}{300}-5.378\times10^{-5}
= 0.0033333-0.00005378
= 0.0032795\ {\rm K^{-1}}.$$
Taking the reciprocal to obtain $$T_2$$,
$$T_2 = \frac{1}{0.0032795}\approx 304.92\ {\rm K}.$$
The required rise in temperature is therefore
$$\Delta T = T_2 - 300 = 304.92 - 300 \approx 4.92\ {\rm K}.$$
Hence, the correct answer is Option A.