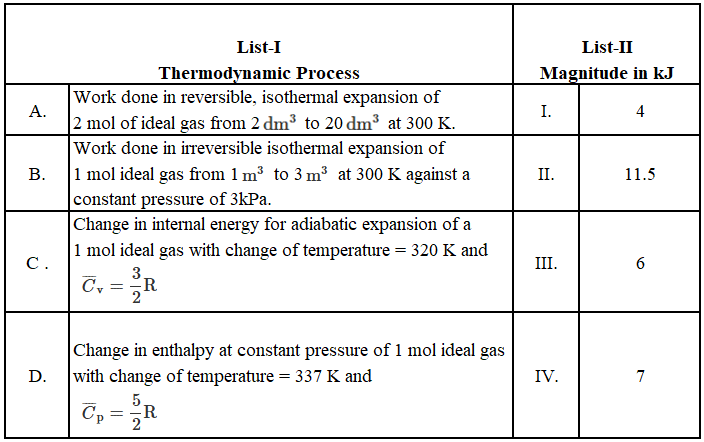
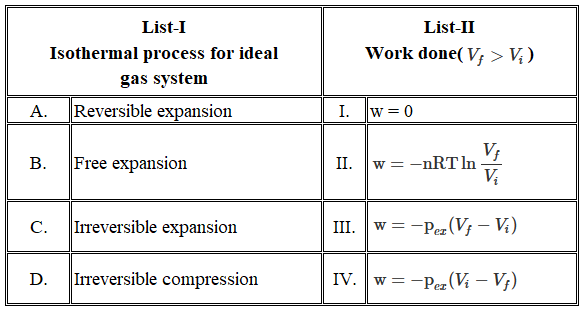
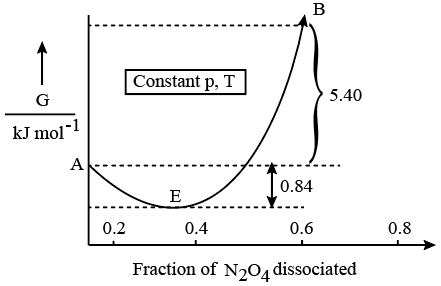
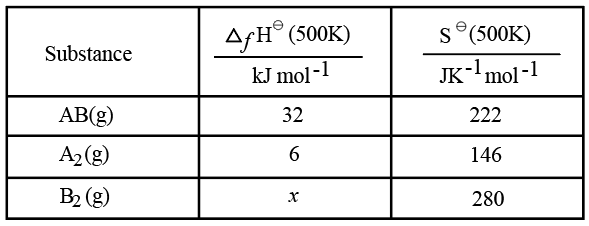
We have an acid-base neutralisation taking place between the protons supplied by H$$_2$$SO$$_4$$ and the hydroxide ions supplied by NaOH. First, we calculate the number of moles present in each solution.
The molarity-volume relation is stated as $$n = M \times V$$, where $$n$$ is moles, $$M$$ is molarity in mol L$$^{-1}$$ and $$V$$ is volume in litres.
For the sulphuric acid solution, the volume is 400 mL = 0.400 L and the molarity is 0.2 M, so
$$n(\text{H}_2\text{SO}_4) = 0.2 \,\text{mol L}^{-1} \times 0.400 \,\text{L} = 0.080 \,\text{mol}.$$
Each molecule of H$$_2$$SO$$_4$$ furnishes two protons. Thus, the total moles of H$$^+$$ available are
$$n(\text{H}^+) = 0.080 \,\text{mol} \times 2 = 0.160 \,\text{mol}.$$
For the sodium hydroxide solution, the volume is 600 mL = 0.600 L and the molarity is 0.1 M, so
$$n(\text{NaOH}) = 0.1 \,\text{mol L}^{-1} \times 0.600 \,\text{L} = 0.060 \,\text{mol}.$$
Each NaOH molecule gives one hydroxide ion, hence
$$n(\text{OH}^-) = 0.060 \,\text{mol}.$$
The neutralisation reaction is
$$\text{H}^+(aq) + \text{OH}^-(aq) \rightarrow \text{H}_2\text{O}(l) \qquad \Delta_rH = -57.1 \,\text{kJ mol}^{-1}.$$
Because the stoichiometry is 1 : 1, the limiting reagent is the species present in the smaller amount. We compare:
$$n(\text{H}^+) = 0.160 \,\text{mol} \quad\text{and}\quad n(\text{OH}^-) = 0.060 \,\text{mol}.$$
Clearly $$0.060 \,\text{mol} < 0.160 \,\text{mol},$$ so OH$$^-$$ is the limiting reagent. Therefore, only 0.060 mol of neutralisation can occur.
The heat released, $$q$$, is calculated from the enthalpy change per mole:
$$q = n(\text{reacted}) \times |\Delta_rH| = 0.060 \,\text{mol} \times 57.1 \,\text{kJ mol}^{-1}.$$
Converting to joules (1 kJ = 1000 J),
$$q = 0.060 \times 57.1 \times 1000 \,\text{J} = 3426 \,\text{J}.$$
This heat raises the temperature of the combined solution. The total volume after mixing is
$$V_{\text{total}} = 400 \,\text{mL} + 600 \,\text{mL} = 1000 \,\text{mL} = 1.000 \,\text{L}.$$
The density of water is given as 1.0 g cm$$^{-3}$$ (or 1.0 g mL$$^{-1}$$), so the mass of the final solution is
$$m = 1000 \,\text{mL} \times 1.0 \,\text{g mL}^{-1} = 1000 \,\text{g}.$$
The specific heat capacity, $$c$$, is 4.18 J K$$^{-1}$$ g$$^{-1}$$. The temperature rise $$\Delta T$$ is obtained from the calorimetric relation $$q = m c \Delta T$$, therefore
$$\Delta T = \frac{q}{m c} = \frac{3426 \,\text{J}}{1000 \,\text{g} \times 4.18 \,\text{J g}^{-1}\text{K}^{-1}}.$$
Simplifying the denominator first,
$$m c = 1000 \times 4.18 = 4180 \,\text{J K}^{-1}.$$
Now dividing,
$$\Delta T = \frac{3426}{4180} \,\text{K} = 0.820 \,\text{K (to three significant figures)}.$$
We rewrite this temperature change in the form $$x \times 10^{-2}$$ K:
$$0.820 \,\text{K} = 82.0 \times 10^{-2} \,\text{K}.$$
Rounding 82.0 to the nearest integer gives 82.
So, the answer is $$82$$.